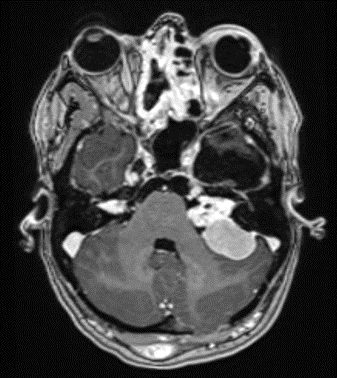
Hintergrund Genetik und klinische Ausprägung Neurofibromatose Typ 2 (NF2) ist eine autosomal-dominat vererbte Erkrankung, die durch multiple Tumoren des zentralen und peripheren Nervensystems sowie Läsionen der Augen und der Haut charakterisiert ist (1;2). Die Prävalenz liegt bei 1:60.000, die Inzidenz bei etwa 1:33.000 (1:25.000 bis 1:87.000) Geburten (3-6). Das betroffene Gen auf dem Chromosom 22q12 wurde 1993 von Arbeitsgruppen in Quebec und Boston identifiziert (7;8). Es kodiert ein Tumorsupressorprotein, welches als MERLIN (Moesin-Ezrin-Radixin-Like-Protein) oder Schwannomin bezeichnet wird. Patienten erben entweder eine Keimzellmutation des Allels eines Elternteils oder erkranken durch eine postzygotische Neumutation eines Allels während der Embryogenese. Molekulargenetische Tests werden bestenfalls an frisch entnommenem Tumorgewebe bei der Entfernung des ersten Tumors durchgeführt (9;10). Das ubiquitäre Vorkommen von MERLIN in nahezu allen Zelltypen des Menschen erklärt die vielgestaltige Symptomatik im Falle einer systemischen Mutation (Keimbahnmutation). Am häufigsten entwickeln sich bilaterale Vestibularisschwannome (VS, Abb. 1). Intrakranielle Meningeome und Tumore im Wirbelkanal und Rückenmark sind ebenfalls häufig, ebenso wie Schwannome der peripheren Nerven und anderer Hirnnerven. Entgegen der Benennung der Erkrankungen treten dabei Neurofibrome – Tumore der Nervenfasern – sehr selten auf. Vorherrschend sind Schwannome (Synonym: Neurinome), also Tumore der Nervenscheiden (Schwann-Zellen). Persistierende Reparaturvorgänge durch Schwann-Zellen in einem veränderten axonalen Mikromilieu tragen wahrscheinlich zur Schwannomentstehung bei (11).
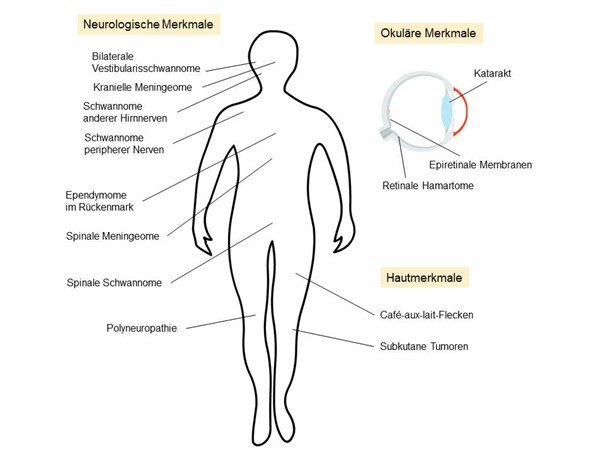
Die NF2 bleibt zunächst eine klinische Diagnose (Abb. 2), auch wenn die genetischen Untersuchungen gut etabliert und in den meisten Fällen treffsicher sind (12). Die WHO hat bisher keine eigene Systematik der NF2-Kriterien publiziert, allerdings gibt es einen sehr detaillierten diagnostischen Score von Baser et al. aus dem Jahr 2011 (13). Die Inzidenz der drei klinischen Hauptkriterien (neurologische, okuläre, dermatologische Manifestationen) variiert in der Literatur zum Teil erheblich (1).
Seit der grundlegenden Unterscheidung der Ausprägung in Wishart- und Gardner-Typ haben mehrere Untersuchungen inzwischen eine Beziehung zwischen Phänotyp und Genotyp der Erkrankung dokumentiert. Wie oben beschrieben, führen alle Abbruch-Mutationen (Nonsense oder Frameshift) zu einem schwereren Verlauf der Erkrankung (14-18). Deletionen mit konsekutivem vollständigem Verlust des Proteinprodukts MERLIN und Missense-Mutationen sind mit milderen Verläufen assoziiert. Splice-Mutationen sind mit unterschiedlicher Krankheitsausprägung verbunden. Betreffen sie die Exons 1-5 sind die Verläufe meist schwerer als bei Mutationen in den Exons 11-15 (19;20). Damit verbunden ist offenbar auch die Lebenserwartung der Patienten (21). Mosaike beeinflussen den Phänotyp ganz entscheidend. Dabei steht der Anteil der von der Mutation betroffenen somatischen Zellen in Zusammenhang mit dem klinischen Verlauf der Erkrankung: Patienten mit einer geringeren Anzahl betroffener Zellen werden voraussichtlich eine mildere Ausprägung zu erwarten haben, zum Teil mit asymmetrischer (in Bezug auf die Tumorlast in den Kleinhirnbrückenwinkeln) oder oligolokaler Erkrankung z. B. rein im Kleinhirnbrückenwinkel gelegener oder zumindest rein intrakranieller Lokalisation (22). Trotz der o. g. Studien, die eine enge Verknüpfung zwischen Geno- und Phänotyp nahelegen, ist dieser Zusammenhang zwischen Mutationstyp und Mosaikbildung ganz sicher nicht linear. Die klinischen Krankheitsverläufe variieren beträchtlich, besonders aber bezüglich des Wachstumsverhaltens einzelner Tumoren (23). Besonders eindrucksvoll dokumentiert sich das bei den pathognomonischen Vestibularisschwannomen (24), deren Wachstumsverhalten völlig unabhängig von der Gesamttumorlast sein kann (25). Entscheidend für ein sinnvolles Timing von Interventionen bezüglich NF2-assoziierter Tumoren ist die Kenntnis des Wachstumsverhaltens der Tumoren. Retrospektive Studien anhand des im NF-Zentrum Erfurt behandelten Patientenkollektivs konnte entscheidend zum Erkenntnisgewinn auf diesem Gebiet beitragen. Das Wachstum der Tumore variiert sowohl nach Tumorart und Lokalisation (26;27) als auch nach individuellen molekulargenetischen Eigenschaften der Tumore (6;28;29). Intrakraniell zeigen Vestibularisschwannome und Meningeome ähnliche durchschnittliche jährliche Wachstumsraten zwischen 0.30 und 2.57cm3 (26;30-33). Nicht-vestibuläre Schwannome hingegen wachsen im Erwachsenalter sehr langsam und bleiben oft über mehrere Jahre größenkonstant (23;34). Den drei genannten Tumorentitäten ist gemeinsam, dass das häufigste Wachstumsmuster eine „saltatorische“ Größenzunahme ist (23;26). Das bedeutet, dass auf Wachstumsperioden Wachstumspausen folgen. Unsere volumetrischen Untersuchungen ergaben für intrakranielle VS und Meningeome ein medianes progressionsfreies Intervall von 18-21 Monaten (23;35). Behandlungsergebnisse müssen vor dem Hintergrund dieser biologischen Wachsstumsunterbrechungen beurteilt werden. Spinale intramedulläre NF2-assoziierte Tumore sind fast ausnahmslos Ependymome (36;37). In zwei Studien mit nicht-volumetrischen Messungen wuchsen intramedulläre Tumore entweder gar nicht (bei 9 Patienten über im Mittel 77 Monate) bzw. nur in 27 % der Fälle (bei 3 von 11 Patienten über im Mittel 52 Monate (38;39). In einer am Neurofibromatosezentrum Erfurt durchgeführten volumetrischen Studie lag das progressionsfreie Intervall im Median bei 24 Monaten (40). 71 % der Tumoren wuchsen über ein medianes Intervall von 76,5 Monaten, 19 % blieben stabil und 10 % schrumpften. Wieder war ein saltatorisches Wachstumsmuster vorherrschend (73 %), gefolgt von linearem Wachstum (18 %). Ein exponentielles Wachstum wurde bei diesen Tumoren nicht beobachtet. 5 (11,9 %) der beobachteten Tumore mussten im Verlauf reseziert werden. Das jährliche Wachstum intraduraler extramedullärer Tumoren (v. a. Meningeome und Schwannome) Wachstum liegt im Mitttel deutlich unter 10 % (41;42). Management bei der Erstvorstellung in einer Neurofibromatose-Ambulanz oder in einem entsprechenden Zentrum werden die Schwerpunkte auf folgende Punkte gesetzt:
- eine ausführliche Anamnese
- familiäre Aspekte und genetische Stammbäume
- eine physische Untersuchung fokussiert auf neurologische und Hautmerkmale
- eine Untersuchung durch einen mit der Neurofibromatose vertrauten Augenarzt
- eine audiologische Untersuchung mit Reinton- und Sprachaudiogramm und eine MRT der gesamten Neuroachse
- ein Ganzkörper-MRT-Scan als Ausgangsbefund kann sinnvoll sein
Bei Patienten mit intrakraniellen Tumoren empfiehlt sich eine erneute kranielle MRT-Diagnostik 6 Monate nach dem Erstbefund. Sind die Tumore dann stabil, erfolgt jährlich eine MRT des Kopfes. Das gleiche gilt für größere Tumoren des Rückenmarkskanals. Wenn keine spinale Manifestation nachgewiesen wurde, dann sollte eine erneute spinale Bildgebung spätestens bei Auftreten einer entsprechenden Symptomatik erfolgen. Bei Nachweis kleiner und mittelgroßer spinaler Tumoren ist meist eine Routine-Nachuntersuchung nach 2-3 Jahren ausreichend. Wenn sich das Tumorwachstum nach dem 20. Lebensjahr verlangsamt, können Kontrollintervalle mit Bildgebung von 3 Jahren ausreichend sein. Bei sehr stabilen, milden Verläufen ist in einzelnen Fällen eine weitere Verlängerung der Intervalle ab dem 40. Lebensjahr möglich (43). Klinische Kontrollen schließen augenärztliche apparative Untersuchungen, Audiogramme und Vestibularisprüfungen meist in jährlichen Abständen ein, wobei man hier sicher individuell und symptombezogen variieren kann. Vorteilhaft ist eine langfristige Betreuung der Patienten an einer interdisziplinär aufgestellten Einrichtung – nach Möglichkeit geführt durch wenige medizinische Ansprechpartner, welche die Patienten und deren medizinische und soziale Situation genau kennen. Außer der genetischen Beratung erscheint in vielen Fällen eine psychotherapeutische Begleitung wichtig, weil die Erkrankung erhebliche Auswirkungen auf das Selbstwertgefühl der Betroffenen und auf die Familiendynamik haben kann (44). Ein kompetentes Management von Patienten mit dem komplexen Krankheitsbild NF2 bedarf eines multidisziplinären Teams (45). Priorität hat die Erhaltung von neuronalen Funktionen, weil damit die Lebensqualität der Patienten untrennbar verbunden ist. Durch Früherkennung, die Betreuung in entsprechenden Zentren und erweiterte therapeutische Möglichkeiten hat sich die Mortalität der Erkrankung kontinuierlich verringert, die mittlere Lebenserwartung liegt jetzt deutlich über 60 Jahren (2;46). Die NF2 ist ein relativ anspruchsvolles Krankheitsbild, nicht nur wegen der fast unausweichlichen Ertaubung der meisten Patienten im Verlauf. Ein kompetentes Management bedarf eines multidisziplinären Teams unter Beteiligung v.a. der Humangenetik, der Neurochirurgie, der Otolaryngologie, der Augenheilkunde, der Neurologie, der Pädiatrie, der Radiologie, der Pathologie, der Strahlentherapie und der Audiologie. Das prioritäre Ziel des Managements muss die Erhaltung von Funktionen sein. Damit untrennbar verbunden ist die Lebensqualität der Patienten. Die Erstmanifestation der NF2 – z.B. eine Mononeuropathie – wird oft noch verkannt. Insbesondere den Neurologen und den Pädiatern kommt daher eine besondere Bedeutung im Rahmen des Managements und der Zuordnung von Frühsymptomen (u.a. Radikulopathien der oberen oder unteren Extremitäten durch extra-axiale spinale Tumoren) zu. In Deutschland werden derzeit NF2-Patienten in einer Vielzahl von neurochirurgischen Kliniken behandelt. Leider fehlt hier teilweise die Kompetenz und Infrastruktur zur optimalen Behandlung der Patienten. Eine Alternative zeigt der Blick über die Landesgrenzen auf: In Großbritannien gibt es nur 4 spezialisierte NF2-Zentren: Addenbrooke’s Hospital in Cambridge, Central Manchester University Hospitals, Guy’s and St Thomas‘ NHS Foundation Trust in London und Oxford University Hospitals Trust. Diese Zentren arbeiten eng zusammen und poolen die Daten ihrer Patienten in einer nationalen Kohorte. Aufgrund der Reduzierung und der damit einhergehenden Spezialisierung der Zentren soll eine effektive und standardisierte Behandlung der Patienten gewähleistet werden. Durch die enge Verlinkung der Zentren ist es möglich, valide Daten aus einem großen Patientenkollektiv zu publizieren (46-48). Expertise des Neurofibromatosezentrums Erfurt Im Orphanet-gelisteten Neurofibromatosezentrum Erfurt ist eine interdisziplinäre spezialisierte Versorgung von Neurofibromatose-Patienten möglich. Mit derzeit über 150 Patienten (Tendenz steigend) behandeln wir eine der deutschlandweit größten NF2-Kohorten. In retrospektiven Studien zum Wachstumsverhalten von NF2-assozzierten Tumoren konnten wir Wachstumsmuster von intrakraniellen und intramedullären spinalen Tumoren identifizieren (23;36;41). Auf Grundlage dieser Daten war es uns möglich, das Wissen über den natürlichen Krankheitsverlauf von NF2 zu erweitern und Empfehlungen für die radiologischen Kontrollintervalle zu formulieren. Zielstellung Mit der Implementierung einer Datenbank für Neurofibromatose-Patienten sind 3 Hauptziele verbunden:
- Vereinfachung der Patientenbehandlung
- Verbesserung der Kommunikation zwischen den deutschsprachigen Neurofibromatose-Zentren
- Generierung hochwertiger prospektiver und wissenschaftlich verwertbarer Daten
Methodik Die Datenbank wurde auf der Castor-EDC-Platform erstellt, welche online zugängig und mit den aktuellen EU-Datenrichtlinien konform ist. Die Inhalte basieren auf der bereits im Memorial Sloan Kettering Hospital in New York verwendeten Datenbank, die uns von Prof. Karajannis zur Verfügung gestellt wurde. Der Einschluss in die Datenbank erfolgt nach schriftlicher Einwilligung der Patienten oder ihrer gesetzlichen Vertreter. In der NF-Registry werden zunächst für jeden Patienten die grundlegenden Einschlusskriterien und unveränderliche Daten (Pseudonym, Genetik, Geschlecht) abgefragt. Anschließend können für jeden ambulanten oder stationären Aufenthalt detaillierte klinische sowie apparative Befunde (u.a. Tumorvolumetrien, audiologische und ophthalmologische Befunde) eingetragen werden. Pseudonymisierte Patiendaten können aus der Datenbank problemlos im PDF- und .csv-Format exportiert werden. Die Datenbank ist auf beliebig viele Studienzentren erweiterbar. Wir planen eine enge Verknüpfung mit anderen Neurofibromatosezentren wie Tübingen und Hamburg. Ausblick Mit den prospektiv gewonnen Daten werden wir verschiedenste Fragen zum Thema Neurofibromatose beantworten können. Dazu gehören:
- Korrelieren Genetik und klinischer Schweregrad?
- Korrelieren Genetik und Wachstumsverhalten der Tumore?
- Welche Langzeiteffekte haben systemische Therapien?
- Wie ist die Lebensqualität der Betroffenen und welche prognostischen Faktoren für die Einschränkung der Lebensqualität gibt es?
- Gibt es geschlechtsspezifische Unterschiede im klinischen Bild oder in Bezug auf Therapeutika?
Literatur (1) Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, Lonser RR. Neurofibromatosis type 2. Lancet 2009 June 6;373(9679):1974-86. (2) Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Harris R. A clinical study of type 2 neurofibromatosis. Q J Med 1992 August;84(304):603-18. (3) Antinheimo J, Sankila R, Carpen O, Pukkala E, Sainio M, Jaaskelainen J. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000; 54(1): 71-6. (4) Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010; 152A(2): 327-32. (5) Evans DG, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005; 26(1): 93-7. (6) Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009; 4: 16. (7) Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, . Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993 June 10;363(6429):515-21. (8) Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K, . A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993 March 12;72(5):791-800. (9) Dumanski JP, Carlbom E, Collins VP, Nordenskjold M. Deletion mapping of a locus on human chromosome 22 involved in the oncogenesis of meningioma. Proc Natl Acad Sci U S A 1987 December;84(24):9275-9. (10) Seizinger BR, Rouleau G, Ozelius LJ, Lane AH, St George-Hyslop P, Huson S, Gusella JF, Martuza RL. Common pathogenetic mechanism for three tumor types in bilateral acoustic neurofibromatosis. Science 1987 April 17;236(4799):317-9. (11) Schulz A, Buttner R, Hagel C, Baader SL, Kluwe L, Salamon J, et al. The importance of nerve microenvironment for schwannoma development. Acta Neuropathol. 2016; 132(2): 289-307. (12) Mautner VF, Lindenau M, Kaufmann D. Klinik und Genetik der Neurofibromatose. Dt Aerzteblatt 1995;92(24):A1759-A1764. (13) Baser ME, Friedman JM, Joe H, Shenton A, Wallace AJ, Ramsden RT, et al. Empirical development of improved diagnostic criteria for neurofibromatosis 2. Genet Med. 2011; 13(6): 576-81. (14) Evans DG, Trueman L, Wallace A, Collins S, Strachan T. Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): evidence for more severe disease associated with truncating mutations. J Med Genet 1998 June;35(6):450-5. (15) Baser ME, Kuramoto L, Joe H, Friedman JM, Wallace AJ, Gillespie JE, Ramsden RT, Evans DG. Genotype-phenotype correlations for nervous system tumors in neurofibromatosis 2: a population-based study. Am J Hum Genet 2004 August;75(2):231-9. (16) Kluwe L, Bayer S, Baser ME, Hazim W, Haase W, Funsterer C, Mautner VF. Identification of NF2 germ-line mutations and comparison with neurofibromatosis 2 phenotypes. Hum Genet 1996 November;98(5):534-8. (17) Parry DM, MacCollin MM, Kaiser-Kupfer MI, Pulaski K, Nicholson HS, Bolesta M, Eldridge R, Gusella JF. Germ-line mutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet 1996 September;59(3):529-39. (18) Ruttledge MH, Andermann AA, Phelan CM, Claudio JO, Han FY, Chretien N, Rangaratnam S, MacCollin M, Short P, Parry D, Michels V, Riccardi VM, Weksberg R, Kitamura K, Bradburn JM, Hall BD, Propping P, Rouleau GA. Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet 1996 August;59(2):331-42. (19) Baser ME, Kuramoto L, Woods R, Joe H, Friedman JM, Wallace AJ, Ramsden RT, Olschwang S, Bijlsma E, Kalamarides M, Papi L, Kato R, Carroll J, Lazaro C, Joncourt F, Parry DM, Rouleau GA, Evans DG. The location of constitutional neurofibromatosis 2 (NF2) splice site mutations is associated with the severity of NF2. J Med Genet 2005 July;42(7):540-6. (20) Kluwe L, MacCollin M, Tatagiba M, Thomas S, Hazim W, Haase W, Mautner VF. Phenotypic variability associated with 14 splice-site mutations in the NF2 gene. Am J Med Genet 1998 May 18;77(3):228-33. (21) Bianchi AB, Hara T, Ramesh V, Gao J, Klein-Szanto AJ, Morin F, Menon AG, Trofatter JA, Gusella JF, Seizinger BR, . Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nat Genet 1994 February;6(2):185-92. (22) Evans DG, Ingham SL. Reduced life expectancy seen in hereditary diseases which predispose to early-onset tumors. Appl Clin Genet 2013;6:53-61. (23) Lawson McLean AC, Rosahl SK. Growth dynamics of intracranial tumors in patients with neurofibromatosis type 2. World Neurosurgery. 2017; 98: 152-61. (24) Fisher LM, Doherty JK, Lev MH, Slattery WH. Concordance of bilateral vestibular schwannoma growth and hearing changes in neurofibromatosis 2: neurofibromatosis 2 natural history consortium. Otol Neurotol 2009 September;30(6):835-41. (25) Baser ME, Makariou EV, Parry DM. Predictors of vestibular schwannoma growth in patients with neurofibromatosis Type 2. J Neurosurg 2002 February;96(2):217-22. (26) Dirks MS, Butman JA, Kim HJ, Wu T, Morgan K, Tran AP, et al. Long-term natural history of neurofibromatosis Type 2-associated intracranial tumors. J Neurosurg. 2012; 117(1): 109-17. (27) Evers S, Verbaan D, Sanchez E, Peerdeman S. 3D Volumetric Measurement of Neurofibromatosis Type 2-Associated Meningiomas: Association Between Tumor Location and Growth Rate. World Neurosurg. 2015; 84(4): 1062-9. (28) Dewan R, Pemov A, Kim HJ, Morgan KL, Vasquez RA, Chittiboina P, et al. Evidence of polyclonality in neurofibromatosis type 2-associated multilobulated vestibular schwannomas. Neuro Oncol. 2015; 17(4): 566-73. (29) Heineman TE, Evans DG, Campagne F, Selesnick SH. In Silico Analysis of NF2 Gene Missense Mutations in Neurofibromatosis Type 2: From Genotype to Phenotype. Otol Neurotol. 2015; 36(5): 908-14. (30) Abaza MM, Makariou E, Armstrong M, Lalwani AK. Growth rate characteristics of acoustic neuromas associated with neurofibromatosis type 2. Laryngoscope. 1996; 106(6): 694-9. (31) Goutagny S, Bah AB, Henin D, Parfait B, Grayeli AB, Sterkers O, et al. Long-term follow-up of 287 meningiomas in neurofibromatosis type 2 patients: clinical, radiological, and molecular features. Neuro Oncol. 2012; 14(8): 1090-6. (32) Li H, Hao SY, Wang L, Li D, Wu Z, Zhang LW, et al. Factors influencing the growth rate of vestibular schwannoma in patients with neurofibromatosis type 2. Acta Neurochir (Wien ). 2015; 157(11): 1983-90. (33) Nowak A, Dziedzic T, Czernicki T, Kunert P, Morawski K, Niemczyk K, et al. Strategy for the surgical treatment of vestibular schwannomas in patients with neurofibromatosis type 2. Neurol Neurochir Pol. 2015; 49(5): 295-301. (34) Fisher LM, Doherty JK, Lev MH, Slattery WH, III. Distribution of nonvestibular cranial nerve schwannomas in neurofibromatosis 2. Otol Neurotol. 2007; 28(8): 1083-90. (35) Lawson MA, Rosahl S. Gender-specific growth dynamics of neurofibromatosis type-2-related tumors of the central nervous system. Acta Neurochir (Wien ). 2016; 158(11):2127-34. (36) Lee M, Rezai AR, Freed D, Epstein FJ. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996; 38(1): 32-7. (37) Mautner VF, Tatagiba M, Lindenau M, Funsterer C, Pulst SM, Baser ME, et al. Spinal tumors in patients with neurofibromatosis type 2: MR imaging study of frequency, multiplicity, and variety. AJR Am J Roentgenol. 1995; 165(4): 951-5. (38) Dow G, Biggs N, Evans G, Gillespie J, Ramsden R, King A. Spinal tumors in neurofibromatosis type 2. Is emerging knowledge of genotype predictive of natural history? J Neurosurg Spine. 2005; 2(5): 574-9. (39) Rennie AT, Side L, Kerr RS, Anslow P, Pretorius P. Intramedullary tumours in patients with neurofibromatosis type 2: MRI features associated with a favourable prognosis. Clin Radiol. 2008; 63(2): 193-200. (40) Lawson McLean AC, Rosahl SK. Growth dynamics of intramedullary spinal tumors in patients with neurofibromatosis type 2. Clin Neurol Neurosurg. 2016; 146: 130-7. (41) Lee CH, Chung CK, Hyun SJ, Kim CH, Kim KJ, Jahng TA. A longitudinal study to assess the volumetric growth rate of spinal intradural extramedullary tumour diagnosed with schwannoma by magnetic resonance imaging. Eur Spine J. 2015; 24(10): 2126-32. (42) Ozawa H, Onoda Y, Aizawa T, Nakamura T, Koakutsu T, Itoi E. Natural history of intradural-extramedullary spinal cord tumors. Acta Neurol Belg. 2012; 112(3): 265-70. (43) Lloyd SK, Evans DG. Neurofibromatosis type 2 (NF2): diagnosis and management. Handb Clin Neurol. 2013; 115: 957-67. (44) Mautner VF, Lindenau M, Kaufmann D. Klinik und Genetik der Neurofibromatose. Dt Aerzteblatt. 1995; 92(24): A1759-A1764. (45) Blakeley JO, Evans DG, Adler J, Brackmann D, Chen R, Ferner RE, et al. Consensus recommendations for current treatments and accelerating clinical trials for patients with neurofibromatosis type 2. Am J Med Genet A. 2012; 158A(1): 24-41. (46) Hexter A, Jones A, Joe H, Heap L, Smith MJ, Wallace AJ, et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: a UK national analysis of 1192 patients. J Med Genet. 2015; 52(10): 699-705. (47) Morris, K.A., Golding, J.F., Blesing, C. et al. Toxicity profile of bevacizumab in the UK Neurofibromatosis type 2 cohort. J Neurooncol. 2017; 131: 117-24. (48) Morris, K.A., Golding, J.F, Axon,P.F., et al. Bevacizumab in neurofibromatosis type 2 (NF2) related vestibular schwannomas: a nationally coordinated approach to delivery and prospective evaluation. Neuro Oncol Practice. 2016; 3(4): 28-9.